Block copolymers as a structured material
A mesoscopic model of a diblock copolymer is used to study the formation, stability, and coarsening of lamellar phases. With the transition to new technologies in fields such as micro electronics or biomedicine, microstructural control of the polymer is necessary in order to meet particular performance requirements. As a consequence, increased emphasis has been placed on research on microstructure and self-assembly at the mesoscale. We mention, for example, the use of block copolymers as nanolithographic templates, or as photonic band gap materials, light emitting diodes and dielectric mirrors. The central challenge for widespread application in both areas is to achieve long-ranged domain order of the appropriate orientation.
 Diblock copolymers are macromolecules comprising two chemically distinct
and mutually incompatible segments (monomers) that are covalently bonded.
Their equilibrium properties are dictated by N, the degree or polymerization
(i.e., the length of the chain), f, the volume fraction of one of the
monomers, and Χ, the Flory-Huggins interaction
parameter between the
distinct segments. The first two parameters can be controlled through
processing, whereas the third is determined by the choice of monomers involved.
Diblock copolymers are macromolecules comprising two chemically distinct
and mutually incompatible segments (monomers) that are covalently bonded.
Their equilibrium properties are dictated by N, the degree or polymerization
(i.e., the length of the chain), f, the volume fraction of one of the
monomers, and Χ, the Flory-Huggins interaction
parameter between the
distinct segments. The first two parameters can be controlled through
processing, whereas the third is determined by the choice of monomers involved.
Above the order-disorder transition temperature TODT,
the equilibrium phase is fluid like (disordered) (below, left).
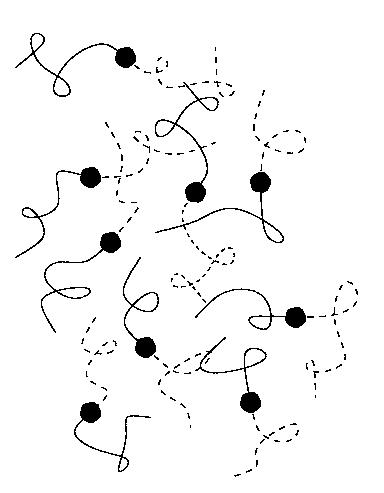 Below TODT, equilibrium structures of a
wide variety of symmetries have been predicted and experimentally observed.
Around f=0.5 (symmetric mixture), a so called lamellar phase is
observed (below, right), in which nanometer sized
layers of A and B rich regions alternate in space.
Below TODT, equilibrium structures of a
wide variety of symmetries have been predicted and experimentally observed.
Around f=0.5 (symmetric mixture), a so called lamellar phase is
observed (below, right), in which nanometer sized
layers of A and B rich regions alternate in space.
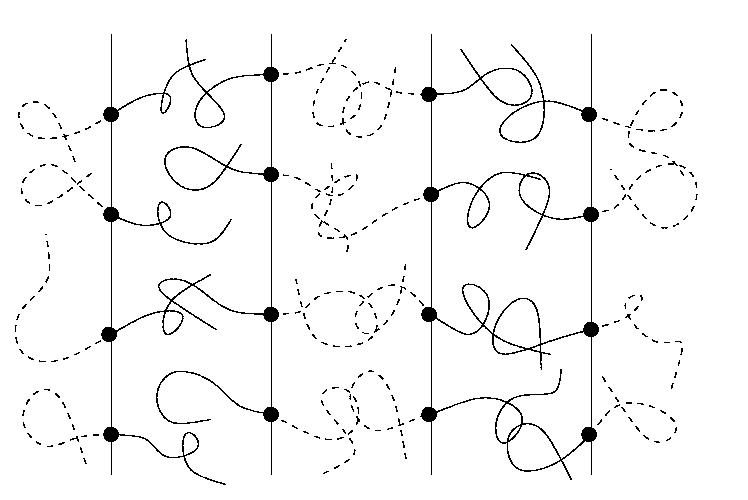
Processing by temperature quench or solvent casting from a high temperature isotropic phase always leads to the formation of a polycrystalline structure comprised of domains or grains of differently oriented lamellae (see figure below). In practice, full development of the equilibrium structure (a single domain or grain of uniform orientation) requires processing times which are at least as long as the time required for substantial ordering at the scale of the size of the system; yet the underlying ordering rates of block copolymers are essentially unknown at present. Not only partial ordering is detrimental for some applications, but it can also lead to aging of the material, as well as to potentially anomalous response to applied stresses.
Orientation by reciprocating shear flows
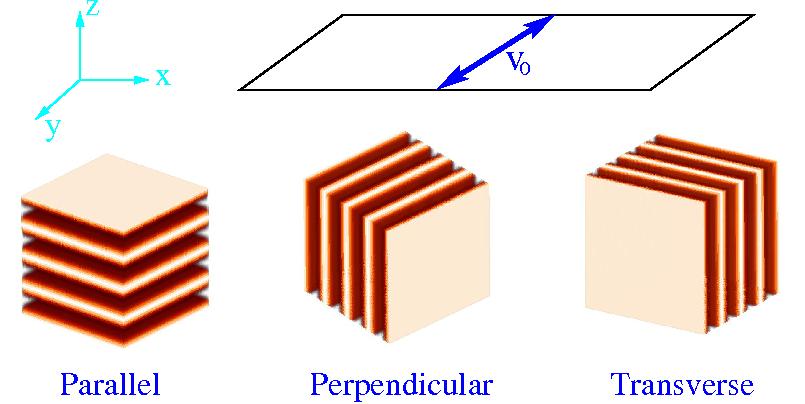
Several methods are currently in use to accelerate the development of long-ranged order of block copolymer microstructures. We have focused on one the most common for bulk samples: an externally imposed reciprocating shear as shown in the figure.
In practice, however, a variety of lamellar orientations are observed depending on the architecture of the block and the parameters of the shear, while the mechanisms responsible for orientation selection are not yet understood. For the purposes of the discussion, it is convenient to classify ordered configurations according to the three possible orientations relative to the shear:
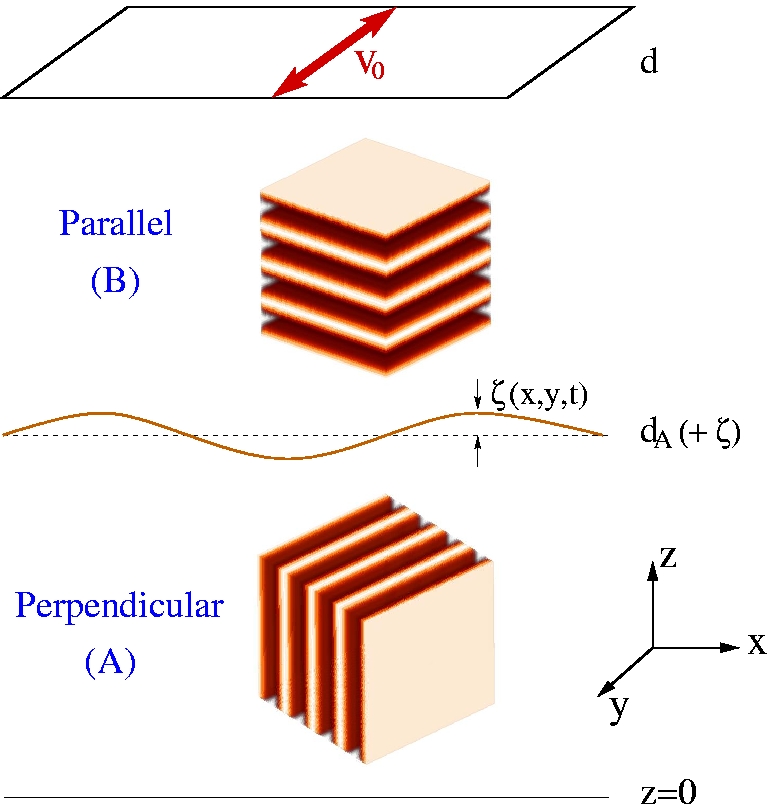
We have been able to determine the regions of stability of each
orientation as a function of the parameters of the shear. The figure
shows a particular example as a function of lamellar orientation.
A perfect lamellar structure is linearly stable within the toroidal
region shown.
 A configuration in the unstable region will decay to a different pattern
within the stable region.
The following animation shows a lamellar pattern under a low
frequency shear flow that is unstable to a transverse perturbation.
The structure becomes unstable, forms a series of "kink bands" as
an intermediate stage, and finally re-orients at a different angle relative to
the shear (MPEG file, 1879 kB, calculation by Francois
Drolet).
A configuration in the unstable region will decay to a different pattern
within the stable region.
The following animation shows a lamellar pattern under a low
frequency shear flow that is unstable to a transverse perturbation.
The structure becomes unstable, forms a series of "kink bands" as
an intermediate stage, and finally re-orients at a different angle relative to
the shear (MPEG file, 1879 kB, calculation by Francois
Drolet).
Unfortunately this stability diagram is not sufficient to explain orientation selection as observed in the experiments, as all orientations retain a certain range of stability. Our recent research has been exploring dynamical selection mechanisms based on the motion of grain boundaries that separate regions of differently oriented lamellae.
 The experimentally relevant range of shear frequencies is well below the
inverse characteristic relaxation times of the polymer chains, and hence a
reduced description in terms of the monomer volume fraction is adopted.
According to this description, the lamellar phase response
is solid like or elastic for perturbations directed along the lamellae normal,
and fluid like or viscous
on the lamellar plane. In the limit of vanishing frequency, the viscous part of
the response has been assumed to be Newtonian with uniform shear viscosity,
and therefore the experimentally relevant parallel and perpendicular
orientations are degenerate and unmodified by the shear.
Our current research addresses the consequences of what we believe is the
leading deviation away from Newtonian response in the limits of low frequency
and characteristic flow scale much longer than the lamellar spacing: the
viscous stress tensor of a lamellar phase is, by reason of symmetry, the same
as that of any other uniaxial phase (e.g., a nematic liquid crystal).
The slowly varying local wavevector of the lamellae
plays a role analogous to that of the director in a nematic.
The experimentally relevant range of shear frequencies is well below the
inverse characteristic relaxation times of the polymer chains, and hence a
reduced description in terms of the monomer volume fraction is adopted.
According to this description, the lamellar phase response
is solid like or elastic for perturbations directed along the lamellae normal,
and fluid like or viscous
on the lamellar plane. In the limit of vanishing frequency, the viscous part of
the response has been assumed to be Newtonian with uniform shear viscosity,
and therefore the experimentally relevant parallel and perpendicular
orientations are degenerate and unmodified by the shear.
Our current research addresses the consequences of what we believe is the
leading deviation away from Newtonian response in the limits of low frequency
and characteristic flow scale much longer than the lamellar spacing: the
viscous stress tensor of a lamellar phase is, by reason of symmetry, the same
as that of any other uniaxial phase (e.g., a nematic liquid crystal).
The slowly varying local wavevector of the lamellae
plays a role analogous to that of the director in a nematic.
We have considered the case of an interface separating two domains oriented parallel and perpendicular to the shear, and shown that this configuration is hydrodynamically unstable. The resulting secondary flows break the degeneracy between parallel and perpendicular lamellar orientation, leading to a preferred perpendicular orientation in certain ranges of parameters of the polymer and of the shear. Our predictions rely on the new viscoelastic model of the polymer introduced, and as such as still awaiting experimental confirmation.